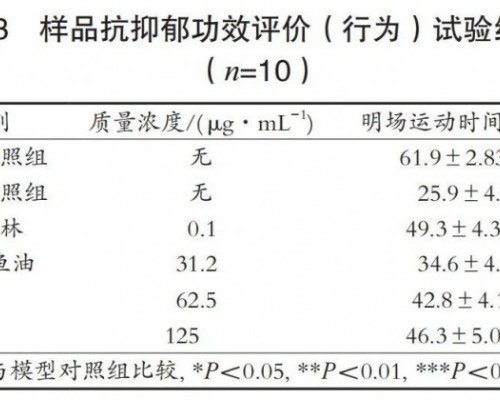
遗传性视网膜营养障碍(RD)是一类遗传异质性疾病,体现为黄斑区光感受器细胞变性、丢掉及视网膜头绪膜萎缩。中心葡萄膜炎(IU)是累及睫状体平整部、玻璃体基底部、周边视网膜及头绪膜的一种炎性、增殖性疾病,在儿童中发病率较高。RD 以进行性视杆视锥功能障碍及变性为临床特征,IU 以双眼眼内炎(首要发生于玻璃体及睫状体平整部)为特征,但二者发病初期不体现典型特征,且均随同黄斑囊样水肿(CME),对确诊形成很大困扰。
为此,Ymkje 等解析了 6 例年青患者(起先被确诊为 IU,后来确诊为 RD),旨在为区别 RD 与 IU 带来一些启示。
患者年纪 5~22 岁,其间,5 位没有宗族视网膜或免疫疾病史,1 位不知道。患者均视力低下(20/25~20/50),1 位有夜盲症。裂隙灯查看显现细微或无眼前节炎症,轻度至中度玻璃体混浊,眼底查看显现均患 CME。无患者呈现 RD 典型色素细胞变性。
患者承受视界测验,成果显现不同程度的周边残缺或环状暗点,光学相干断层扫描(OCT)显现患者均有 CME,黄斑中心凹厚度为 206~651μm,中心视网膜外核层变薄,这指示或许患 RD 或长时刻炎性 CME 引起的黄斑萎缩。4 位患者承受荧光素血管造影术查看,2 位体现为强荧光,2 位为毛细血管渗漏。全视界视网膜电图(ffERG)成果低于正常值,被认定为葡萄膜炎继发。
鉴于上述临床特征,医生确诊患者为 IU,实施眼周及/或眼内皮质类固醇医治后,3 位 CME 暂时改进,4~8 周后复发;4 位患者承受乙酰唑胺医治 ≥ 3 个月,但对 CME 无效;5 位患者承受多重免疫调节疗法,但并未长时刻减轻 CME 及炎症,也未进步视力。
随访时刻 1~8 年不等,患者 CME 继续,视力恶化,3 位呈现夜盲症状,4 位继续性视界残缺,一切患者暗视及光视 ffERG 应对严峻反常。
选用微阵列芯片法检测患者是否存在基因骤变,成果显现,3 位患者 CRB1 位点骤变(2 位骤变相同,具有潜在致病性),1 位 RP1 位点骤变,1 位 USH2A 位点骤变,2 位显性 RD 基因骤变,1 位患者与家人失掉联络,显性遗传无法证明,但其母亲曾患夜盲症。
综大将患者确诊为 RD,改动医治办法,患者病况好转。
RD 的正确确诊对医治很重要,可防止大剂量免疫抑制剂的过错运用,削减患者苦楚。但发病初期,因为 RD、IU 均不体现典型临床症状,且存在 CME、类似于 CME 的黄斑反常及视网膜沉积物等类似症状,易导致误诊,如上述病例。
关于 IU,确诊参阅如下:玻璃体炎症反响重,可见雪球样混浊及雪堤样渗出,常伴有视乳头炎、CME、周边视网膜血管炎及后囊下白内障等。关于无痛温文 IU 患者,Ymkje 等以为,若患者体现 CME,但无睫状体平整部炎症,应考虑前期 RD 的或许性,主张医生直接问询有无夜盲症及宗族视网膜疾病史,一起联合视界测验、ffERG 及基因测序等办法进一步确诊,并咨询专门 RD 医生。